|
| |
Projects

|
|
|
Funding
Agencies |
Leveraged
partially from different grants |
|
Program Managers |
|
|
Start date |
(initial
development started on September, 1996)
June 2006 |
|
Expires |
on-going |
| |
|
|
Investigators |
Past collaborators:
 |
Tahir Cagin (now
at Texas A&M) |
|
Amir Fijany
(JPL) |
|
| Abstract |
|
 |
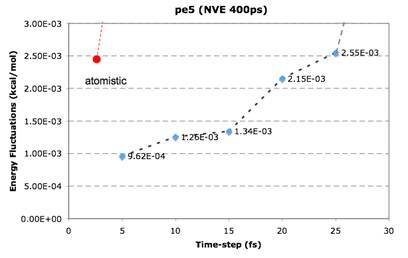
In the study of molecular systems using Molecular Dynamics
(MD) simulations it is often desired to impose relative or absolute motion
constraints on atoms or groups of atoms. This offers several different
advantages depending on the type of constraint involved, i.e. absolute or
relative, and on the type of generalized coordinates used to describe the
equations of motion (EOM) of the system, i.e. Cartesian or internal.
Indistinctively, both approaches involve the formulation of mixed systems of
differential EOM and nonlinear algebraic equations of constraint.
Our approach involves the use of internal coordinates,
even though this increases the complexity of the solution to the EOM
(mainly because we now have a highly coupled molecular system with highly
non-linear, non inertial terms in velocity, a dense mass matrix operator and
potential numerical conditioning of the formulation solution) hence
requiring efficient algorithms to compute the algebraic differential
solution in linear, or better, computation time, it offers several
advantages over the conventional Cartesian constrained dynamics methods (the
Figure to the left depicts an NVT@300K of a polymer chain that results in an
order of magnitude increase in simulation time-step in comparison to
conventional atomistic Cartesian MD).
We have successfully demonstrated its use in large-scale,
long-term dynamics of complex systems in polymer and protein science [see
Publication list]. Current research involves
addressing challenges in closed loop kinematics systems and correcting bad
contact dynamics without time penalties. |
| |
| Related Publications |
| Jaramillo-Botero, A. "Molecular
Nanomanipulator Dynamic Design Criteria" In: Dekker Encyclopedia of
Nanoscience and Nanotechnology. 1st Ed. New York : Marcel Dekker Publ.,
2004. |
| Jaramillo-Botero, A., Matta, A.,
Correa, JF, Perea, W. Software platform for robot modeling and simulation,
Vol. 3, No. 1, December 2004 |
| Jaramillo-Botero, A. "Computational
Nanotechnology in the Design of Nanoscale Molecular Positional Devices",
NASA Jet Propulsion Laboratory Seminar (invited talk), Pasadena,
California (US) November 23, 2004 |
| Jaramillo-Botero, A. and Crespo, A.
"A Unified Formulation For Massively Parallel Rigid Multibody Dynamics Of
O(Log2 N) Computational Complexity", Journal of Parallel and Distributed
Computing, Academic Press, Vol. 62, No. 6, June 1, 2002. |
| Jaramillo-Botero, A "Design
Criteria for a 3 DOF positional nanomanipulator based on a Constrained
Molecular Dynamics Model" In: Mathematics in Nanoscale Science and
Engineering 2004 Reunion Conference (invited talk), 2004, University of
California at Los Angeles, Lake Arowhead Conference, California (US)
January 2004. |
| Massively Parallel Algorithms for
Long-term Simulations of Large-scale Molecular Systems, Universidad
Politecnica de Valencia, Doctoral Thesis, (spanish) ISBN: 958-8162-60-2,
1998. |
| Fijany, A., Jaramillo-Botero, A., Cagin, T., and
Goddard, W.A. III, A Fast Algorithm for Massively Parallel, Long Term
Simulations of Complex Molecular Dynamics Systems, pp 505-515 (1998), in
Parallel Computing: Fundamentals, Applications and New Directions, Eds. E.
H. D'Hollander, G. R. Joubert, F. J. Peters and U. Trottenberg.
|
| Fijany, A., Cagin, T., A., Jaramillo-Botero, and
Goddard, W. A. III. Novel Algorithms for massively parallel, long term
simulation of molecular dynamics systems, Advances in Engineering
Software, 29, 441-450 (1998). |
| Fijany, A., T. Cagin, A. Jaramillo-Botero, S. Gulati,
and W.A. Goddard,"Novel Algorithms for Massively Parallel, Long-Term,
Simulation of Molecular Dynamics Systems," 4th NASA National Symposium on
Large-Scale Analysis and Design on High-Performance Computers and
Workstation, Williamsburg, VA, October 1997. |
| Fijany, A., T. Cagin, A. Jaramillo-Botero, and W.A.
Goddard, "Massively Parallel Constraint Force Algorithm for MD Simulation
of Polymers and Dendrimers," Presented at the 1997 American Physical
Society (APS) Int. Conf. on Computational Physics (PC97), Santa Cruz, CA,
Aug. 1997. |
| Fijany, A. A. Jaramillo -Botero, T. Cagin, and W.A.
Goddard, "A Fast Algorithm for Massively Parallel, Long-Term, Simulation
of Complex Molecular Dynamics Systems," Proceedings Parallel Computing 97
(PARCO 97), Bonn, Germany, September 1997. |
| Fijany, A., T. Cagin, A. Jaramillo -Botero, T. Coley,
and W.A. Goddard, "A Massively Parallel Algorithm for Solution of
Constrained Equations of Motion with Application to Large-Scale Long-Time
Molecular Dynamics Simulations," Presented at the 2nd Parallel
Computational Chemistry Symposium, American Chemistry Society (ACS), San
Francisco, CA, April 1997. |
| Fijany, A., T. Cagin, A. Jaramillo -Botero, and W.A.
Goddard,"A Massively Parallel Algorithm for Solution of Constrained
Equations of Motion in Molecular Dynamics," Presented at the American
Physical Society Meeting, Kansas City, MO, March 1997. |
| Rigid Multibody Molecular Dynamics: Strictly Parallel
Computations, ISBN: 958-33-4988-7, TR-RAG-1996 - Robotics and Automation
Group (RAG), 1996. online: http://ingenieria.puj.edu.co/centros/cap/proyectos/multicuerpos/html/Intro.html |
|
|

