|



| |
Projects

|
|
|
Partnership |
College of Petroleum Engineering
and Geoscience (CPG) at the King Fahd University of Petroleum and Minerals (KFUPM) |
|
Tentative Start date |
2016 |
|
Tentative End date |
2019 |
| |
|
|
People Involved |
|
| Project Summary |
|
 |
| Collaborative
research effort on polymer colloidal science applied to enhanced and
improved oil recovery technologies (EOR/IOR), focused specifically on the
application of unique first-principles based multiscale modeling and
simulation methods, and novel experimental material synthesis and
characterization capabilities, to: understand, validate, steer design, and
optimize synthesis of smart nano-polymeric fluids for EOR/IOR.
The general expected outcomes include:
1. New multiscale simulation tools for simulating complex fluid phenomena
2. New validated materials providing controllable dynamic shear response
3. Improved research & education capacity on complex fluid phenomena at the
CPG
More specifically, this effort will considerably enhance the fundamental
understanding of the atomistic nanoscale mechanisms responsible for
self-induced shear thickening/thinning in “smart” polymer fluid compositions
applicable to EOR/IOR.
The key personnel: Goddard, Jaramillo-Botero, Tang, and Jiang bring
more than 120 years of experience relevant to this project with expertise
spanning fundamental theory, modeling, and experiments on complex polymer
and nanoscale systems. |
| |
| Project Proposal |
|
| Project Summary |
|
| Reports |
 | private |
|
| Related Publications |
| private |
|
| |
|
|
|
|
Funding
Agencies |
NNSA |
| Project |
Caltech Predictive Science
Academic Alliance Program (PSAAP). Lead by Michael Ortiz. (Exec. Dir.
Mark Stalzer) |
|
Start date |
2009 |
|
Expires |
2014 |
| |
|
|
MSC
Investigators and students involved |
|
Patrick
Theofanis |
|
Hai Xiao |
|
Qi An |
|
Chi Feng (SURF
2011) |
|
| Abstract |
|
|
|
|
Our role in
this effort has been on the development of first-principles-based multiscale
methods to understand the dynamics of materials during the extreme
conditions of hypervelocity impact (HVI), including the characterization of
pressure and temperature dependent material transformations from ground
state, through warm-dense regimes and up to plasma phases. A critical goal
for HVI modeling is to capture the non-adiabatic effects on material
properties that result from medium to large electronic excitations, and to
understand the particle interactions that take place between high-energy
electrons, nuclei, ions and larger species during HVI. The warm-dense regime
is particularly challenging since it lays in a "computational no-mans land"
between the cold matter described well by ground-state quantum mechanics
(QM) methods (e.g. density functional theory, DFT) and the hot matter
described well by classical plasma models (e.g. Particle-in-Cell, PIC). We
have developed unique methods to address these issues via mixed
quantum-classical theory, which extend the limited capabilities of QM
calculations into large-scale dynamics (millions of atoms) of systems at
finite (and large) temperatures/pressures. In addition to this fundamental
role, our group provides first-principles predicted properties to
parameterize atomistically-informed continuum methods developed by other
groups in the Caltech-PSAAP effort (e.g. OTM).
 Our
contributions to date have included the development of reaxFF reactive force
fields for accurately modeling the reaction processes and phases of
different materials of interest to NNSA (e.g. Nylon, Al, Ta, Fe), Tight
Binding (TB) models to implicitly correct reaxFF trajectories for
high-energy electronic effects, as well as the
development of a non-adiabatic quantum-classical wave-packet dynamics
method called the electron force field (eFF) for modeling the explicit
dynamics of electronically excited states (Figure to the left shows the
general flow for HVI simulations using our methods). eFF has been
demonstrated and validated for ground state and highly excited system
dynamics [1,3,4,5,7,9-11]. Our
contributions to date have included the development of reaxFF reactive force
fields for accurately modeling the reaction processes and phases of
different materials of interest to NNSA (e.g. Nylon, Al, Ta, Fe), Tight
Binding (TB) models to implicitly correct reaxFF trajectories for
high-energy electronic effects, as well as the
development of a non-adiabatic quantum-classical wave-packet dynamics
method called the electron force field (eFF) for modeling the explicit
dynamics of electronically excited states (Figure to the left shows the
general flow for HVI simulations using our methods). eFF has been
demonstrated and validated for ground state and highly excited system
dynamics [1,3,4,5,7,9-11].
Using ReaxFF we have modeled the
effect of reaction processes, including bond breaking and formation and
phase transformations, in the extreme conditions of temperature and pressure
that occur during the HVI (<10km/s), for materials including Ta, Fe, Al, and
Nylon. To incorporate electronic structure effects in ReaxFF we have tested the use of a TB approach that restricts the basis sets to
the minimal set needed to capture the excitations in the bands of
interest, i.e. it focuses only on the bands near the Fermi energy for a
particular molecular system, to avoid the cost of a complete basis set[13]. The
key benefit of this first-principles parameterized TB approach is that it
can scale to systems with tens of millions of atoms, while the description
remains at the atomic level, with the position of each atom described
explicitly as well as the electronic density at the atomic site.
At higher velocities (>10km/s) we
have shown the applicability of eFF to understand the electronic effects on material properties,
including cascaded electronic ionization, non-adiabatic dissociation,,
increased conductivity, among other phenomena. We have validated the
eFF approach for low-Z elements (H, He, Li, Be, B, and C) using
full electron representations via floating spherical Gaussian wavefunctions
and to overcome the limitations of spherical Gaussians in describing p and
higher order angular momentum orbitals, multiple bonds and lone pairs in eFF,
we introduced first-principles-based effective core potentials (ECP), which
describes the valence electrons in the vicinity of core electrons
combined as a single pseudo core particle. As a result atoms C, N, O, Na, Al, and Si are now described with accurate bonding energy and geometries in the eFF
model. This results in proper description of multiple bonds (sigma-pi rep.)
and lone pairs (open shell rep.), but further efforts are necessary for
accurate representation of conjugation and hydrogen bonding. To reach the length- and time-scales required for studying materials under
extreme conditions, e.g. in Rayleigh-Taylor instabilities, we have made our
parallel implementation
of eFF available as a user package in the Sandia code LAMMPS.
|
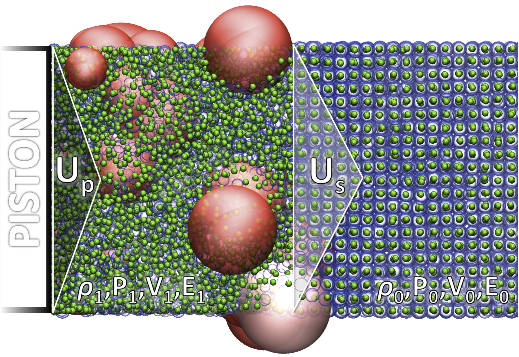 |
We have applied eFF on large-scale,
long-term dynamics problems, including: static and dynamic shock (figure on
the left shows a piston driven at HV into a lithium slab, depicting electron
excitations in the compressed front as red spheres and a
movie of two lithium slabs colliding at HV)
Hugoniots for Lithium [7,11], poly(ethylene) [1],
material interfacial instabilities,
high-pressure EOS for Aluminum and Nylon [results now in use by the Ortiz
group in the OTM hypervelocity impacts of Nylon on Aluminum], brittle
fracture dynamics (Movie shows a Si
crystal delaminating from uniaxial mechanical loading - electronic energies
are color-coded) of silicon[3], and hypervelocity impact of different
molecular species and clathrates [5].
In addition to our force fields
effort in PSAAP, we have developed new methods to improve the accuracy of
DFT-QM and incorporated corrections to describe the London dispersions
responsible for long-range van der Waals attractions in our ReaxFF engine[12].
The first-principles-based approach
provides increased confidence in our simulation results, but to estimate the
level of accuracy we confront our results against available experimental
data and use both results, from computations and experiments, to estimate
the level of uncertainty involved using the Uncertainty Quantification (UQ)
formalisms (Owhadi and Ortiz) and code (Aivazis) under development by other
teams in the Caltech PSAAP.
|
| Related Publications |
- Theofanis, P.L, Jaramillo-Botero,
A., Goddard, W.A. III, Mattsson, T., Thompson, A. "Electron dynamics of
shocked polyethylene crystal", Accepted in PRB, March, 2012
- Qi, A., Zybin, S., Goddard, W.A.,
III, Jaramillo-Botero, A. "Elucidation of the dynamics of hot spot
initiation and chemical reactions at interfaces of highly shocked
materials" Accepted in Phys. Rev. B, August 2011.
- Theofanis, P.L, Jaramillo-Botero,
A., Goddard, W.A. III, "Non-adiabatic study of dynamic electronic effects
during brittle fracture in silicon", Accepted in PRL, December, 2011
- Jaramillo-Botero, A. "Modeling and
simulation of large-scale reactive systems in extreme conditions", invited
talk HPC User Forum, San Diego, September 7, 2011.
- Jaramillo-Botero, A., Cheng, MJ,
Beegle, L., Hodyss, R., Goddard, WA III. "Predicting the molecular
composition of Enceladus' south pole plume after hypervelocity impact with
the Cassini orbiter", Journal article in preparation. Data presented at
the 42nd Lunar and Planetary Science Conference, March 7-11, 2011,
Texas.
- Jaramillo-Botero, A., J. Tahir-Kheli, P. von Allmen,
and W.A. Goddard, Multiscale, multiparadigm modeling for nano systems
characterization and design, in Handbook of nanoscience, engineering, and
technology, W.A. Goddard, et al., Editors. 2012, CRC Press, Taylor &
Francis Group.
- Jaramillo-Botero, A., J.T. Su, A. Qi, and W.A. Goddard,
Large-scale, long-term nonadiabatic electron molecular dynamics for
describing material properties and phenomena in extreme environments.
Journal of Computational Chemistry, 2011. n/a. doi: 10.1002/jcc.21637.
- Theofanis, P.L., A. Jaramillo-Botero, and W.A. Goddard,
Non-Adiabatic Study of Dynamic Electronic Effects During Brittle Fracture
of Silicon. Submitted to PRL, 2011.
- Jaramillo-Botero, A., J.T. Su, and W.A. Goddard, pEFF:
the parallel electron force field for large-scale, long-term nonadiabatic
excited electron dynamics under LAMMPS, 2009, http://lammps.sandia.gov/movies.html
- eff: Pasadena.
- Jaramillo-Botero, A., M.J. Cheng, V. Cvicek, L.W.
Beegle, R. Hodyss, and W.A. Goddard. First-principles-based reactive
atomistic simulations to understand the effects of molecular hypervelocity
impact on Cassini's Ion and Neuratl Mass spectrometer. in 42nd Lunar and
Planetary Science Conference. 2011. The woodlands, Texas.
- Kim, H. and W.A. Goddard, The PBE-lg method of improved
London Dispersion for elements up to Lr (103). To be published in Journal
of Physical Chemistry Letters, 2011.
- L. Liu, Y. Liu, S. Zybin, H. Sun and W.A. Goddard III,
"ReaxFF-lg: Correction of the ReaxFF Reactive Force Field for London
Dispersion, with Applications to the Equations of State for Energetic
Materials", Journal of Physical Chemistry A, 115 (40). pp. 11016-11022.
ISSN 1089-5639
- Jaramillo-Botero, A., Tahir-Kheli,
J., von Allmen, P., Goddard WA III, "Multiscale,
multiparadigm modeling for nano systems characterization and design",
CRC handbook of Nanoscience, Engineering and Technology, 3rd ed. Chapter
29. In press (May 2012).
|
| Reports |
| private |
|
| |
|
|
|
|
|
|
Funding
Agencies |
Under contract
with Advanced Research Associates and US Air Force |
|
Start date |
2008 |
|
Expires |
2009 |
| |
|
|
MSC
Investigators |
|
William A.
Goddard, III (Principal Investigator) |
|
Andres
Jaramillo-Botero (Co-Principal Investigator, Coord.) |
|
Yi Liu
(Co-Principal Investigator) |
|
| Abstract |
|
|
|

Aqueous
Film Forming Foam (AFFF) is the prime fire-fighting agent used for liquid
hydrocarbon fires, it works by forming a water film beneath the foam that
cools the liquid fuel and stops the formation of flammable vapors. This
provides dramatic fire knockdown, an important factor in crash rescue fire
fighting. The Air Force (AF) uses this fire-fighting agent in all Aircraft
Rescue and Fire Fighting vehicles to respond to aircraft fires. The
AF Research Laboratory is interested in determining and clearly
understanding the unique processes of this agent for extinguishing burning
liquid hydrocarbons.
This research is meant to understand and elucidate
fundamental structures and properties of fire-fighting AFFFs, derived from
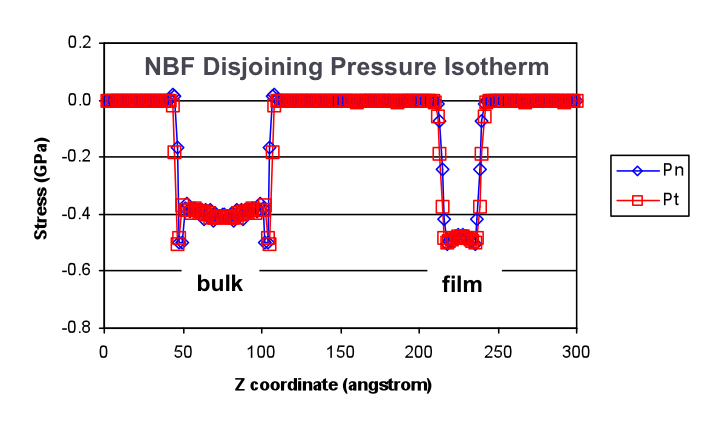
the interfacial systems in Newton-Black Films (Figure to the right shows a
molecular model of a perfluorinated surfactant-based NBF, and figure above
shows a graph of its calculated disjoining pressure isotherm as a measure of
film stability), by leveraging, extending and applying existing
first-principles-based multiscale theory, methods and computational tools
developed at the Materials and Process Simulation Center, at the California
Institute of Technology. These methods and tools will provide accurate,
atomistically resolved, information about AFFFs composition and behavior to
enable Air Force Research Laboratory scientists to predict and optimize
quantities (e.g. reduce volumes) and compositions (e.g. reduce toxicity via
reduction/replacement of fluorinated surfactants) of AFFF precursors for
performance enhancement.
The importance of this work lays on the Air Forces'
requirement for a) advanced technologies that facilitate contingency base
operations and combat support functions, including the development of
environmentally compatible, operational and cost effective fire
extinguishing AFFFs, and b) a reduction in weight, volume, and costs of base
emergency response, infrastructure support, and combat support systems, as
expressed in the corresponding Broad Agency Announcement (BAA). |
| |
| Reports |
| private |
|
| Related Publications |
| private |
|
| |
|
|
|
|
|
|
Start date |
February, 2008 |
|
End date |
July,
2008 |
| |
|
|
People Involved |
|
| Abstract |
|
|
|
 Silicon
(Si) is the
most widely used
semiconductor material in industry. It is fundamental for the production
of semiconductor microelectronic devices as well as to other areas of
application, including, the development of novel high efficiency thin films for solar cells.
Most of these applications require starting with an atomically smooth Silicon surface
which is conventionally achieved using a Chemical Vapor Deposition (CVD)
process to perform epitaxial crystal growth of Si. A model image of the
growth process that takes place during the CVD is shown to the right. Silicon
(Si) is the
most widely used
semiconductor material in industry. It is fundamental for the production
of semiconductor microelectronic devices as well as to other areas of
application, including, the development of novel high efficiency thin films for solar cells.
Most of these applications require starting with an atomically smooth Silicon surface
which is conventionally achieved using a Chemical Vapor Deposition (CVD)
process to perform epitaxial crystal growth of Si. A model image of the
growth process that takes place during the CVD is shown to the right.
In order to understand and optimize the multiscale
properties and phenomena that lead to a smooth semiconductor thin film
surface it is crucial to develop predictive models capable of expressing the
chemical-physical processes involved in crystal growth within a CVD reactor.
 This study involves developing first-principles-based multiscale methods and
tools to
elucidate the influence of adsorbed hydrogen on growing silicon surfaces. It
is known that the presence of hydrogen
adsorbed on the surface can alter the growth regime, leading either to a
desired smooth crystal surface or to an undesired atomically rough surface
morphology. In order to understand the material properties and phenomena of
interest it is imperative to account for critical events that take place at
the atomistic scale, in particular chemical reactions, and their effect on
structure evolution over into the mesoscale length and time scales.
Accurately modeling chemical reactions would ordinarily require quantum
chemical calculations that take place in the femtosecond time-scales,
unfortunately, these are limited to a few hundred atoms (< 1,000 at most)
and understanding the larger scale structural changes over time would
require modeling systems with thousands of atoms over microsecond
time-scales. This study involves developing first-principles-based multiscale methods and
tools to
elucidate the influence of adsorbed hydrogen on growing silicon surfaces. It
is known that the presence of hydrogen
adsorbed on the surface can alter the growth regime, leading either to a
desired smooth crystal surface or to an undesired atomically rough surface
morphology. In order to understand the material properties and phenomena of
interest it is imperative to account for critical events that take place at
the atomistic scale, in particular chemical reactions, and their effect on
structure evolution over into the mesoscale length and time scales.
Accurately modeling chemical reactions would ordinarily require quantum
chemical calculations that take place in the femtosecond time-scales,
unfortunately, these are limited to a few hundred atoms (< 1,000 at most)
and understanding the larger scale structural changes over time would
require modeling systems with thousands of atoms over microsecond
time-scales.
In order to circumvent these limitations we are using a multiscale-multiparadigm approach which
integrates novel first-principles-based reactive force field methods (reaxFF),
to determine the appropriate kinetic rates during diffusion (see movie shown
to the left) and adsorption processes found under experimental conditions of
temperature and pressure, and kinetic
Monte Carlo methods to complete an accurate mesoscale predictive
atomistically-resolved description of the growth process.
|
| |
| Reports |
| private |
|
| Related Publications |
| private |
|
|
|
|
|
|
Funding Agency |
Department of Transportation
(DOT) |
|
Start date |
2010 |
|
End date |
2013 |
| |
|
|
People Involved |
|
| Abstract |
|
|
|
|
Despite
the importance of cement in civil engineering from buildings to bridges to
highways, the fundamental understanding of how the chemistry and composition
of the components in cement play a role in the mechanics and aging property
of the structures remain largely unknown. In particular there is great
controversy about the formation of nanoscale structures in these systems,
upon cement hydration (as a product of the exothermic reactions of water
with the minerals in the cement clinker) and how they play a role in the
expansion processes upon hydration.
Soon after mixing (Portland) cement with water, the
aluminate phase (C3A) mineral in the cement clinker reacts
strongly with the water to form an aluminate-rich gel that in turn reacts
with sulfate in solution to form nano-crystals of ettringite. This phase of
hydration is strongly exothermic and it is often known as the Aft phase
("Alumina, Ferric oxide, tri-sulfate" or Al2O3 – Fe2O3 – tri). The alite and
belite minerals in the cement start to hydrate after the Aft phase has
initiated, forming calcium silicate hydrate and calcium hydroxide, and
leading to an increase in concrete strength. During a period of heat
evolution that lasts for hours, the cement grains react from the surface
inwards, breaking the anhydrous particles into smaller grains. A Ferrite (C4AF)
hydration reaction phase also starts quickly as water is added, but slows
down as a byproduct iron hydroxide gel layer forms on the surface of the
ferrite to inhibit further reactions.
We using our novel reactive force field technologies in
the simultaneous study of the chemistry, structure, mechanics and dynamics
in cement pastes and related materials. In particular, we our focus (at this
stage) is on the properties of ettringite ((CaO)6(Al2O3)(SO3)3·32H2O
or simply C6AS3H32), because of its role in
the long-term deterioration of concrete structures exposed to sulfate attack
and delayed ettringite formation. The formation of ettringite crystals in
the cured matrix of C-S-H causes expansion and cracking of structures. There
are three main hypothesis for the expansion of the cement structure: 1)
crystal growth of ettringite causes disruption, 2) ettringite crystals of
colloidal dimensions imbibe water, creating osmotic pressure, 3) expansion
is not related to the formation of ettringite but to the changes in the
properties of the cement gel. |
| |
| Reports |
| private |
|
| Related Publications |
| private |
|
| |
|
|

