|



| |
Projects

|
|
|
Funding |
Leveraged
partially from different grants |
|
Start date |
(initial
development started on September, 1996)
June 2006 |
|
Expires |
on-going |
| |
|
|
Investigators |
Past collaborators:
 |
Tahir Cagin (now
at Texas A&M) |
|
Amir Fijany
(JPL) |
|
| Abstract |
|
 |
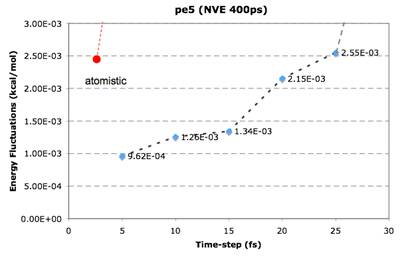
In the study of molecular systems using Molecular Dynamics
(MD) simulations it is often desired to impose relative or absolute motion
constraints on atoms or groups of atoms. This offers several different
advantages depending on the type of constraint involved, i.e. absolute or
relative, and on the type of generalized coordinates used to describe the
equations of motion (EOM) of the system, i.e. Cartesian or internal.
Indistinctively, both approaches involve the formulation of mixed systems of
differential EOM and nonlinear algebraic equations of constraint.
Our approach involves the use of internal coordinates,
even though this increases the complexity of the solution to the EOM
(mainly because we now have a highly coupled molecular system with highly
non-linear, non inertial terms in velocity, a dense mass matrix operator and
potential numerical conditioning of the formulation solution) hence
requiring efficient algorithms to compute the algebraic differential
solution in linear, or better, computation time, it offers several
advantages over the conventional Cartesian constrained dynamics methods (the
Figure to the left depicts an NVT@300K of a polymer chain that results in an
order of magnitude increase in simulation time-step in comparison to
conventional atomistic Cartesian MD).
We have successfully demonstrated its use in large-scale,
long-term dynamics of complex systems in polymer and protein science [see
Publication list]. Current research involves
addressing challenges in closed loop kinematics systems and correcting bad
contact dynamics without time penalties. |
| |
| Related Publications |
| Jaramillo-Botero, A. "Molecular
Nanomanipulator Dynamic Design Criteria" In: Dekker Encyclopedia of
Nanoscience and Nanotechnology. 1st Ed. New York : Marcel Dekker Publ.,
2004. |
| Jaramillo-Botero, A., Matta, A.,
Correa, JF, Perea, W. Software platform for robot modeling and simulation,
Vol. 3, No. 1, December 2004 |
| Jaramillo-Botero, A. "Computational
Nanotechnology in the Design of Nanoscale Molecular Positional Devices",
NASA Jet Propulsion Laboratory Seminar (invited talk), Pasadena,
California (US) November 23, 2004 |
| Jaramillo-Botero, A. and Crespo, A.
"A Unified Formulation For Massively Parallel Rigid Multibody Dynamics Of
O(Log2 N) Computational Complexity", Journal of Parallel and Distributed
Computing, Academic Press, Vol. 62, No. 6, June 1, 2002. |
| Jaramillo-Botero, A "Design
Criteria for a 3 DOF positional nanomanipulator based on a Constrained
Molecular Dynamics Model" In: Mathematics in Nanoscale Science and
Engineering 2004 Reunion Conference (invited talk), 2004, University of
California at Los Angeles, Lake Arowhead Conference, California (US)
January 2004. |
| Massively Parallel Algorithms for
Long-term Simulations of Large-scale Molecular Systems, Universidad
Politecnica de Valencia, Doctoral Thesis, (spanish) ISBN: 958-8162-60-2,
1998. |
| Fijany, A., Jaramillo-Botero, A., Cagin, T., and
Goddard, W.A. III, A Fast Algorithm for Massively Parallel, Long Term
Simulations of Complex Molecular Dynamics Systems, pp 505-515 (1998), in
Parallel Computing: Fundamentals, Applications and New Directions, Eds. E.
H. D'Hollander, G. R. Joubert, F. J. Peters and U. Trottenberg.
|
| Fijany, A., Cagin, T., A., Jaramillo-Botero, and
Goddard, W. A. III. Novel Algorithms for massively parallel, long term
simulation of molecular dynamics systems, Advances in Engineering
Software, 29, 441-450 (1998). |
| Fijany, A., T. Cagin, A. Jaramillo-Botero, S. Gulati,
and W.A. Goddard,"Novel Algorithms for Massively Parallel, Long-Term,
Simulation of Molecular Dynamics Systems," 4th NASA National Symposium on
Large-Scale Analysis and Design on High-Performance Computers and
Workstation, Williamsburg, VA, October 1997. |
| Fijany, A., T. Cagin, A. Jaramillo-Botero, and W.A.
Goddard, "Massively Parallel Constraint Force Algorithm for MD Simulation
of Polymers and Dendrimers," Presented at the 1997 American Physical
Society (APS) Int. Conf. on Computational Physics (PC97), Santa Cruz, CA,
Aug. 1997. |
| Fijany, A. A. Jaramillo -Botero, T. Cagin, and W.A.
Goddard, "A Fast Algorithm for Massively Parallel, Long-Term, Simulation
of Complex Molecular Dynamics Systems," Proceedings Parallel Computing 97
(PARCO 97), Bonn, Germany, September 1997. |
| Fijany, A., T. Cagin, A. Jaramillo -Botero, T. Coley,
and W.A. Goddard, "A Massively Parallel Algorithm for Solution of
Constrained Equations of Motion with Application to Large-Scale Long-Time
Molecular Dynamics Simulations," Presented at the 2nd Parallel
Computational Chemistry Symposium, American Chemistry Society (ACS), San
Francisco, CA, April 1997. |
| Fijany, A., T. Cagin, A. Jaramillo -Botero, and W.A.
Goddard,"A Massively Parallel Algorithm for Solution of Constrained
Equations of Motion in Molecular Dynamics," Presented at the American
Physical Society Meeting, Kansas City, MO, March 1997. |
| Rigid Multibody Molecular Dynamics: Strictly Parallel
Computations, ISBN: 958-33-4988-7, TR-RAG-1996 - Robotics and Automation
Group (RAG), 1996. online: http://ingenieria.puj.edu.co/centros/cap/proyectos/multicuerpos/html/Intro.html |
|
|
|
|
Funding
Agencies |
NNSA-DOE |
|
Start date |
June 2007 |
|
Expires |
on-going |
| |
|
|
Investigators |
|
| Graduate students |
| Qi An (Materials Science) |
| Hai Xiao (Materials Science) |
| Patrick Theofanis (Chemistry) |
|
| Abstract |
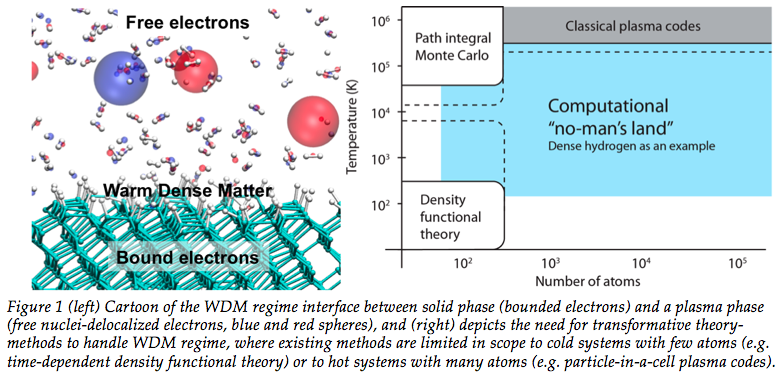 Our research involves developing and
using first principles based methods to examine the chemical processes of
matter at extreme conditions that may have significant concentrations of
molecules, atoms, ions, and electrons exposed to transient mechanical,
thermal and electrical fields. In particular we want to understand the
processes for heterogeneous systems and interfaces in systems with
temperatures ranging from ~0.1-100’s eV [~103-106’sK] and densities up to 10
times standard value (often referred to as warm-dense-matter - WDM - see
top-left figure). Studies
of such systems have mainly been the province of physics and astrophysics.
We want to bring in the chemistry to bridge the transition regimes from
solid to liquid to plasma driven by extreme conditions, using a
first-principles-based modeling and simulation methodology capable of
accurately expressing the non-adiabatic energetics and dynamics of excited
electronic states. For this, we have been developing the
first-principles-based electron force field (eFF) methodology, which enables
modeling of the simultaneous dynamics of electrons and nuclei (eMD) evolving
nonadiabatically under transient extreme conditions. In eFF the N-electron
wavefunction is described as a product of one-electron Gaussian functions,
whose size is a dynamical variable and whose position is not constrained to
a nuclear center. This form allows for straightforward propagation of the
wavefunction, with time, using a simple formulation. The nuclei are
described as point charges. The full Hamiltonian has then a standard
description for electrostatic interactions between a set of delocalized
point and Gaussian charges which include, nuclei-nuclei, electron-electron,
and nuclei-electron. In addition to the electrostatics, eFF introduces QM
effects through an electron kinetic energy from the Gaussians and a
spin-dependent Pauli repulsion potential term between Gaussians. Our research involves developing and
using first principles based methods to examine the chemical processes of
matter at extreme conditions that may have significant concentrations of
molecules, atoms, ions, and electrons exposed to transient mechanical,
thermal and electrical fields. In particular we want to understand the
processes for heterogeneous systems and interfaces in systems with
temperatures ranging from ~0.1-100’s eV [~103-106’sK] and densities up to 10
times standard value (often referred to as warm-dense-matter - WDM - see
top-left figure). Studies
of such systems have mainly been the province of physics and astrophysics.
We want to bring in the chemistry to bridge the transition regimes from
solid to liquid to plasma driven by extreme conditions, using a
first-principles-based modeling and simulation methodology capable of
accurately expressing the non-adiabatic energetics and dynamics of excited
electronic states. For this, we have been developing the
first-principles-based electron force field (eFF) methodology, which enables
modeling of the simultaneous dynamics of electrons and nuclei (eMD) evolving
nonadiabatically under transient extreme conditions. In eFF the N-electron
wavefunction is described as a product of one-electron Gaussian functions,
whose size is a dynamical variable and whose position is not constrained to
a nuclear center. This form allows for straightforward propagation of the
wavefunction, with time, using a simple formulation. The nuclei are
described as point charges. The full Hamiltonian has then a standard
description for electrostatic interactions between a set of delocalized
point and Gaussian charges which include, nuclei-nuclei, electron-electron,
and nuclei-electron. In addition to the electrostatics, eFF introduces QM
effects through an electron kinetic energy from the Gaussians and a
spin-dependent Pauli repulsion potential term between Gaussians.
Thus eFF is a simplified QM method
rather than a conventional force field method, in which electron motions are
averaged out into ground state nuclear motions (i.e a single electronic
state) described by empirically parameterized interatomic potential
functions.
We have demonstrated the applicability
of eFF to multiple problems, including: predicting the single shock
Hugoniots for Hydrogen [Su and Goddard, 2007], Lithium [Kim, Su, and
Goddard, 2010; Jaramillo-Botero, Su, An, and Goddard, 2010], Carbon [An, Su,
Jaramillo-Botero, and Goddard, 2010], Beryllium, Auger dynamics [Su and
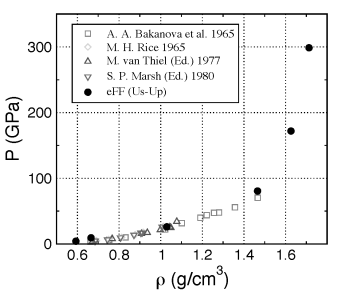
Goddard, 2010], among others. Bottom figure shows the shock wave
kinematics, including mass velocities (Up) and shock wave velocities (Us) ,
and the corresponding single shock Hugoniot for lithium metal, obtained from
a pEFF dynamics simulation.
Current effort focused on:
|
making it practical to
perform simulations of the nonadiabatic dynamics of materials in extreme
environments involving millions of nuclei and electrons, over multi-picosecond
time-scales (for this we have developed pEFF [Jaramillo-Botero et al,
submitted JCC, 2010], see
lithium shock
hypervelocity impact movie), |
|
extend the scope of
our current method (Z=1-6) to include higher p- and d-block elements of
the periodic table (e.g. use of floating elliptical Gaussians to improve
the representation of electrons with p-character), |
|
Improve its efficiency
and accuracy by supporting effective core pseudo potentials, electron
correlation and dispersion |
|
|
|
| |
| Related Publications |
| Carbon Phases Under Extreme Conditions of Temperature
and Pressure during Shock Compression, An, Q; Su, JT; Jaramillo-Botero, A;
Zybin, S; Goddard, WA. In preparation 2010. |
| Large-scale, Long-term Non-adiabatic Electron Molecular
Dynamics for Describing Material Properties and Phenomena in Extreme
Environments, Jaramillo-Botero, A; Su, JT; An, Q; Goddard, WA. in print JCC, 2010. |
| Discovery of New Amorphous Lithium under High
Temperature & High Pressure, Kim, H; Su, JT; Goddard, WA. Under review. |
| The dynamics of highly excited electronic systems:
Applications of the electron force field, Su, JT; Goddard, WA, J. Chem.
Phys. 131 (24): 244501 (2009) |
| Mechanisms of Auger-induced chemistry derived from wave
packet dynamics, Su JT, Goddard WA, P Natl. Acad. Sci. USA 106
(4)1001-1005 (2009) |
| Excited electron dynamics modeling of warm dense
matter, Su JT, Goddard WA, Phys. Rev. Lett. 99 (18): Art. No. 185003
(2007) |
|
|

